发布时间:2020-12-28
附件:紫外治疗设备注册技术审查指导原则(2017年第199号).doc
本指导原则旨在指导和规范紫外治疗设备的技术审评工作,帮助审查人员增进对该类产品机理、结构、主要性能、预期用途等方面的理解,方便审查人员在产品注册技术审评时把握基本的要求和尺度。
本指导原则是对紫外治疗设备的一般要求,申请人应依据产品的具体特性确定其中内容是否适用,若不适用,需具体阐述理由及相应的科学依据,并依据产品的具体特性对注册申报资料的内容进行充实和细化。

本指导原则不作为法规强制执行,不包括行政审批要求。本指导原则所确定的核心内容是在目前的科技认识水平和现有产品技术基础上形成的,因此,审评人员应注意其适宜性,密切关注适用标准及相关技术的最新进展,考虑产品的更新和变化。
一、适用范围
本指导原则适用的紫外治疗设备,是指利用紫外线的物理性能,对人体进行照射治疗的设备(波长范围在200nm—400nm以内),按第二类医疗器械管理。
本指导原则不包括光固化机、紫外激光设备、紫外光敏治疗设备、紫外血液内照射设备、体腔内照射设备。
利用紫外线并结合其他物理方式进行治疗的医疗器械,其紫外线治疗部分亦适用本指导原则。
二、技术审查要点
(一)产品名称的要求
产品的命名应符合《医疗器械通用名称命名规则》(国家食品药品监督管理总局令第19号)和国家标准、行业标准中的通用名称要求,一般由一个核心词和不超过三个特征词组成。紫外治疗设备的核心词一般为治疗仪或光疗仪,特征词为紫外,如:紫外线治疗仪、紫外光疗仪、紫外治疗仪等。
产品名称中不应包括产品型号、系列。
(二)产品的结构和组成
紫外治疗设备一般包括主机部分、辐照器部分、嵌入式软件及其他附属部分,如护目镜或眼罩。主机部分包括机箱、电源模块、控制单元、显示模块。辐照器部分含有紫外光源, 注册申请人应说明光源的型号规格和来源,并提供紫外光源的发射光谱图。申请人应提供护目镜的来源及相关技术参数。
紫外治疗设备按电源部分结构可分为:交流、直流和交直流两用。
紫外治疗设备按照照射人体的部位可分为:全身照射式设备和局部照射式设备。如图1和图2所示。
紫外治疗设备按照可携带形式可分为:台式、立式和便携式设备。
紫外治疗设备按照光源类型可分为:LED光源、荧光光源。
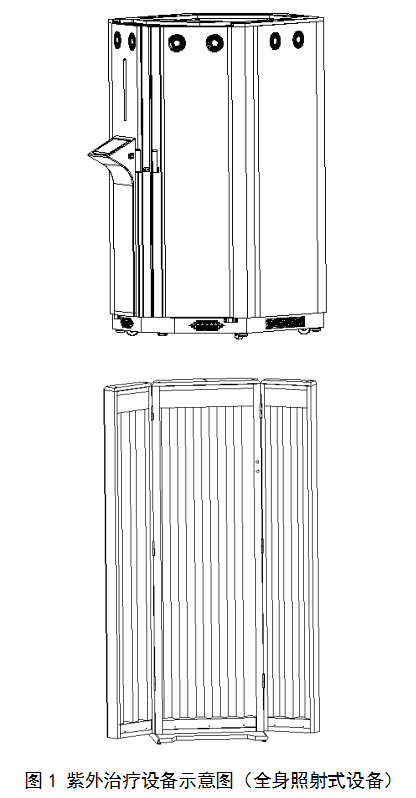
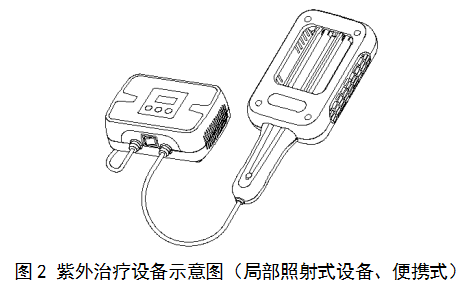
(三)产品工作原理/作用机理
工作原理:紫外治疗设备的辐照装置中装有一个或多个光源,工作时,辐照装置中的光源发出紫外光,用于对患者全身或局部进行照射,控制器根据患者需要照射的剂量来控制紫外线光照时间。一般来说,治疗设备带有散热系统和安全联锁装置。
目前医疗用紫外线分为三段,即长波紫外线UVA(波长320nm—400nm)、中波紫外线UVB(波长275nm—320nm)和短波紫外线UVC(波长275nm—200nm)。
紫外治疗设备常用于治疗银屑病、白癜风。申请人应说明产品的作用机理。若申请人声称可以治疗其他疾病,应提出治疗疾病的机理,注明依据。
(四)注册单元划分的原则和实例
紫外治疗设备的注册单元原则上以产品的技术原理、结构组成、性能指标和适用范围为划分依据。
1.不同电气结构应作为不同注册单元进行注册。
如电击防护类型分别为I类和Ⅱ类的两种设备,应按照两个注册单元进行注册。
若产品采用三相供电与单相供电,则不能放入同一注册单元。
2.不同机械结构,应考虑划分为不同的注册单元。
如全身照射式设备、局部照射式设备;台式、立式和便携式设备按各自分类归入不同的注册单元。
3.光源类型不同,应考虑划分为不同的注册单元。
如采用LED灯和荧光灯做光源的两种设备,应考虑划分为不同的注册单元。
(五)产品适用的相关标准
紫外治疗设备根据产品自身特点适用以下相关标准:
表1 相关产品标准
| 标准编号 | 标准名称 |
| GB 9706.1—2007 | 医用电气设备 第1部分:安全通用要求 |
| GB/T 14710—2009 | 医用电器环境要求及试验方法 |
| GB/T 16886.1—2011 | 医疗器械生物学评价 第1部分:风险管理过程中的评价与试验 |
| GB/T 16886.5—2003 | 医疗器械生物学评价 第5部分:体外细胞毒性试验 |
| GB/T 16886.10—2005 | 医疗器械生物学评价 第10部分:刺激与迟发型超敏反应试验 |
| YY 0901—2013 | 紫外治疗设备 |
| YY 0505—2012 | 医用电气设备 第1—2部分:安全通用要求 并列标准:电磁兼容要求和试验 |
| YY/T 0316—2016 | 医疗器械 风险管理对医疗器械的应用 |
| YY/T 0708—2009 | 可编程医用电气系统 |
| YY/T 0709—2009 | 医用电气设备 第1—8部分:安全通用要求 并列标准:通用要求,医用电气设备和医用电气系统中报警系统的测试和指南 |
| GB 4706.85—2008 | 家用和类似用途电器的安全 紫外线和红外线辐射皮肤器具的特殊要求 |
上述标准包括了技术要求中经常涉及到的部件标准和方法标准。有的申请人还会根据产品的特点引用一些行业外的标准和一些较为特殊的标准......

站点声明:
本网站所提供的信息仅供参考之用,并不代表本网赞同其观点,也不代表本网对其真实性负责。图片版权归原作者所有,如有侵权请联系我们,我们立刻删除。如有关于作品内容、版权或其它问题请于作品发表后的30日内与本站联系,本网将迅速给您回应并做相关处理。
北京飞速度医疗科技有限公司专注于医疗器械、诊断试剂产品政策与法规规事务服务,提供产品注册申报代理、临床合同(CRO)研究、产品研发、GMP质量辅导等方面的技术外包服务。
ONE-STOP SERVICE
免费赠送3万家医疗器械企业名录









